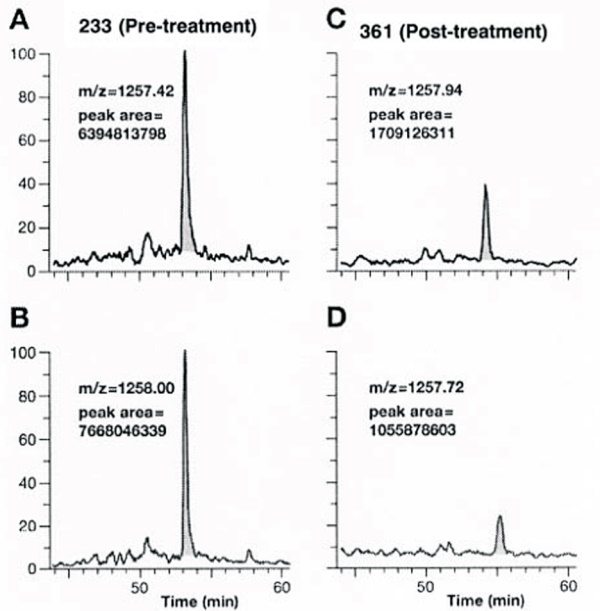
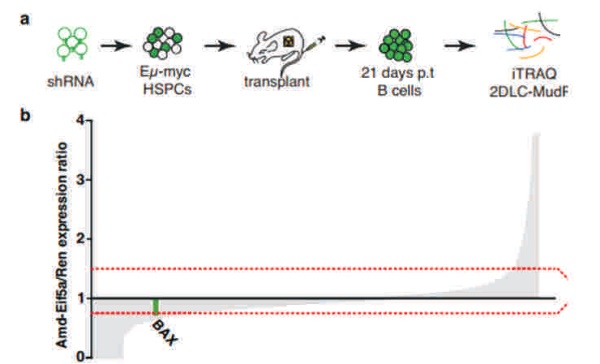
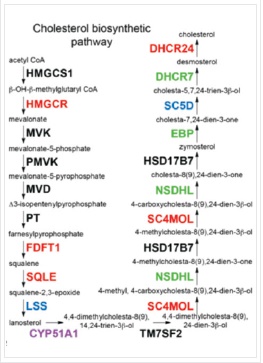
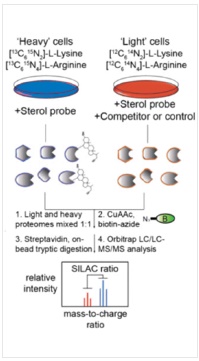
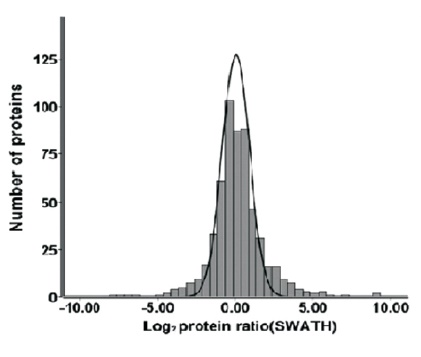
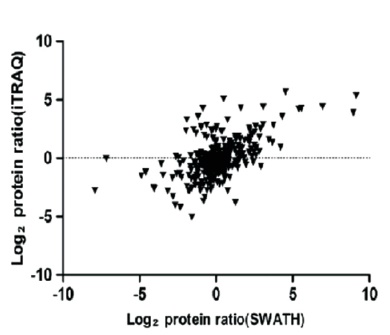
The proteomics content shared in the last issue, is it that some small partners are still in the fog? This time, Xiaobian speaks with examples to help you sort out the details. First, Label-free quantification Label-free quantification , as the name implies, does not require specific labeling of the comparative sample. It is only necessary to compare the chromatographic mass spectrometry response signals of specific peptides/proteins between different samples to obtain the change in protein expression between samples, which is usually used for analysis. Mass spectrometry data generated during scale protein identification and quantification. Label-free technology can be divided into two methods based on Spectra Count (SC) and peptide-based parent ion intensity (or XIA of chromatographic ion current), which is more accurate and widely used. Technical characteristics No need to mark, easy to operate; Not limited by the number of comparative samples; High requirements for stability and repeatability of experimental operations; The accuracy is worse than the mark; At least three technical repetitions or biological replicates are required. Scope of application Suitable for quantitative comparison of large sample sizes; Easy to use label-free technology for experimental designs that cannot be quantified with markers; Classic Case Title: Label-free global serum proteomic profiling reveals novel celecoxib-modulated proteins in familial adenomatous polyposis patients (Label-free quantitative technique for whole-genome analysis of serum from patients with familial adenomatous polyposis, looking for Celecoxib regulatory proteins) Journal: Cancer genomics proteomics Main technology: Label-free quantification Abstract: Celecoxib is a selective cyclooxygenase inhibitor that can prevent and treat patients with familial adenomatous polyposis (FAP) and sporadic colorectal cancer. In order to identify its specific mechanism of action, the multi-dimensional chromatographic separation method combined with electrospray ionization tandem mass spectrometry was used to compare the proteome expression of serum samples before and after treatment with Celecoxib. A significant difference in expression levels of 83 possible expressions for Celecoxib was found. Some of the Celecoxib regulatory proteins were further confirmed in the FAP patients and colorectal cancer cell lines by Western blotting, which laid a foundation for further large-scale clinical trials of the mechanism of Celecoxib. Figure: Comparison of quantitative results of characteristic peptides in Apo A-II protein Note: AB is before processing, CD is processed Fatima N, Chelius D et al. Label-free global serum proteomic profiling reveals novel celecoxib-modulated proteins in familial adenomatous polyposis patients. Cancer genomics proteomics. Source: Fatima N, Chelius D, Luke BT, Yi M, Zhang T, Stauffer S, Stephens R, Lynch P, Miller K, Guszczynski T et al: Label-free global serum proteomic profiling reveals novel celecoxib-modulated proteins in familial Adenomatous polyposis patients. Cancer genomics proteomics 2009, 6(1): 41-49. Second, iTRAQ quantification The iTRAQ (isobaric tags for relative and absolute quantitation) technique is also the most widely used technique in quantitative proteomics. The iTRAQ reagent is divided into a reporter group, a balance group, and a peptide reactive group. There are 8 reporting groups (113, 114, 115, 116, 117, 118, 119, 121 Da). The peptide reactive group can be covalently linked to the N-terminus and the lysine side chain of the peptide chain, thereby marking the reporter group and the balanced group on the peptide; the mass of the equilibrium part is 192-184, respectively, and is added to the report part. Just 305. In the first-order mass spectrum, any one of the iTRAQ reagents labeled the same protein in different samples exhibited the same mass-to-charge ratio. In the secondary mass spectrometry, the equilibrium group in the iTRAQ reagent is neutrally lost, and the signal ions appear as peaks with different mass-to-charge ratios (113-121). According to the height and area of ​​the peak, the same protein can be obtained in different samples. The difference in expression; at the same time, the MS/MS results of the peptides combined with the database search can identify the corresponding protein species. Technical characteristics High sensitivity: detection of lower abundance proteins, cytoplasmic proteins, membrane proteins, nuclear proteins, extracellular proteins, etc.; Strong separation ability: can separate acid/basic protein, protein less than 10K or more than 200K, poorly soluble protein; High throughput: 8 samples can be analyzed simultaneously, especially for differential protein analysis using multiple treatments or samples from multiple treatment times; The results are reliable and accurate: qualitative and quantitative simultaneously, and the identification and quantitative results are obtained. High degree of automation: liquid and liquid use, automated operation, fast analysis, good separation. Scope of application Suitable for any type of sample; Mark up to 8 samples at a time; Classic Case Title: A tumour suppressor network relying on the polyamine-hypusine axis (a novel tumor suppression network based on the Polyamine-hypusine axis) Journal: Nature Main technology: iTRAQ quantification Abstract: In order to find the tumor suppressor gene of lymphoma, we first identified some genes that inhibit tumor expansion in mouse lymphoma model by screening short hairpin RNA library. Then using iTRAQ quantification technology to study the expression regulation of two significant lymphoma tumor suppressor genes, AMD1 and Eif5A, and found that both genes are associated with Hypusine (Hypusine is a unique amino acid, a highly conserved pathway The resulting polyamine metabolites are related. Through further secondary screening, a novel tumor suppressor gene network of the polyamine Hypusine axis was identified, which regulates apoptosis. Figure: iTRAQ quantification process and protein ratio distribution Scuoppo C, Miething C et al. A tumour suppressor network relying on the polyamine-hypusine axis. Nature. Source: Scupop C, Miething C, Lindqvist L, Reyes J, Ruse C, Appelmann I, Yoon S, Krasnitz A, Teruya-Feldstein J, Pappin D et al: A tumour suppressor network relying on the polyamine-hypusine axis. Nature2012 , 487 (7406): 244-248. Third, SILAC quantification SILAC (Stable Isotope Labeling Strategies) provides an effective solution for comprehensive, systematic qualitative and quantitative analysis of complex mammalian cell proteomes. The basic principle of SILAC is to replace the corresponding amino acids in the cell culture medium with essential amino acids labeled with natural isotope (light) or stable isotope (neutral or heavy), and the cells are stably mixed with isotopically labeled amino acids after 5-6 doubling cycles. The original amino acid is replaced by a newly synthesized protein into the cell. The lysed proteins of different labeled cells are mixed in the proportion of cell number or protein, separated and purified, and then identified by mass spectrometry. According to the area comparison of the two isotopic peptides in the first-order mass spectrum, the relative quantitation is carried out, which belongs to the metabolic labeling method in vivo. Technical characteristics High efficiency: SILAC is an in vivo labeling technology with a labeling efficiency of up to 100%; Quantitative accuracy, wide linear range, reduced experimental error due to sample preparation, experimental operation and instrumentation, and good quantitative repeatability; In vivo metabolic markers are combined with SDS-PAGE or chromatographic separation techniques and are compatible with hydrophobic proteins and alkaline proteins, independent of protein properties; Since the technology belongs to the in vivo labeling, the labeling effect is stable, and the labeling efficiency is not affected by the lysing solution, and is not only suitable for the analysis of whole cell proteins, but also for the identification and quantification of membrane proteins; Compared with chemical markers, SILAC method protein requirements are significantly reduced, usually only tens of micrograms of protein per sample; Living mark, closer to the true state of the sample; Compatibility: A variety of proteins expressed in cells or bacteria capable of being cultured in DMEM or RPMI 1640 medium can be labeled. Scope of application It is only suitable for cells cultured in vivo, and cannot be analyzed for tissue samples, body fluid samples, etc. commonly used in biomedical research. Classic Case Title: Proteome-wide Mapping of Cholesterol-Interacting Proteins in Mammalian Cells (Analysis of Cholesterol-Protein Interactions in Mammalian Cells Using a Sterol Probe Combined with SILAC Quantitation Technique) Journal: Nat Methods Main technology: SILAC quantification Abstract: Cholesterol is an important structural component of cell membranes, and it is also a signal molecule of precursors. It plays a major role in the regulation of specific interactions with proteins. In this paper, a clickable photo-sensitive sterol probe combined with SILAC proteomic quantification technology directly resolves cholesterol-protein interactions directly in living cells. More than 250 cholesterol-binding proteins were eventually identified, including receptors, channel proteins, and enzymes involved in many established and previously unreported interactions. The most prominent of the newly discovered protein interactions are the regulation of sugars, glycerides and cholesterol itself, as well as enzymes involved in vesicle trafficking and protein glycosylation and degradation of proteins. These proteins point to biochemical pathways at key nodes, suggesting that sterol concentrations and other metabolite control may be highly correlated with protein localization and modification. Figure: Basic flow of the experiment Figure: Cholesterol biosynthesis pathway Note: Black character protein indicates no identification Jonathan J et al. Proteome-wide Mapping of Cholesterol-Interacting Proteins in Mammalian Cells. Nature methods. Source: Jonathan J, Armand B, Micah J, Sarah E, Benjamin F: Proteome-wide Mapping of Cholesterol-Interacting Proteins in Mammalian Cells.Nature methods, 2013 March; 10(3):259-264. Fourth, SWATH quantification SWATH (Sequential Window Acquisition of all Theoretical Mass Spectra) is a new mass spectrometry acquisition mode technology jointly developed by Dr. Ruedi Aebersold of the Federal Institute of Technology in Zurich, Switzerland, and AB-SCIEX in 2012. It is one of the MS/MSALL technologies. An extension. It divides the scan range into a series of intervals at intervals of 25 Dalton, and uses ultra-high-speed scanning to obtain all fragmentation information of all ions in the scan range. Technical characteristics High sensitivity: SWATH technology inherits the MRM data acquisition mode, combined with the high-resolution AB sciex Triple TOF-plus, with comparable sensitivity to MRM; High reproducibility: the quantitative correlation between repeated samples can reach 0.99 or more; Wide linear dynamic range: the quantitative range can span 4 orders of magnitude; Large flux: More than 2,000 proteins can be detected and quantified in one experiment. Scope of application It is best to detect samples with lower complexity such as bacteria; Can be used in conjunction with MRM technology to discover biomarkers; The interacting proteins can be identified in conjunction with TAP techniques. Classic Case Title: SWATH-and iTRAQ-based quantitative proteomic analyses revealed an overexpression and biological relevance of CD109 in advanced NSCLC (Using SWATH and iTRAQ quantification techniques to reveal overexpression and bio-relationship of CD109 in advanced non-small cell lung cancer) Journal: Journal of proteomics Main technology: SWATH, iTRAQ Abstract: The article uses iTRAQ and SWATH quantitative techniques to analyze the expression changes of protein in high-metastatic and low-metastasis non-small cell lung cancer cell lines (compared with normal state). 110 and 71 proteins with significant differences were found in the cell line secreted protein group and non-small cell lung cancer serum, respectively. The researchers found that among these differential proteins, CD109 has a higher expression level in the highly metastatic cell line secretome and late cancer patients, and the same conclusion was obtained by ELISA. In addition, CD109 is also highly expressed in lung cancer cells as compared to normal lung cells. The researchers also found that knocking out CD109 protein affects the biological function of NSCLC cells, such as inhibiting cell growth and affecting the cell cycle. These phenomena suggest that CD109 plays an important role in the process of NSCLC and is likely to be a key biomarker for lung cancer. Figure: SWATH quantitative protein expression ratio distribution iTRAQ quantification and SWATH quantitative protein ratio correlation distribution Ophthalmic Surgical Pack,Disposable Surgical Opthalmic Pack,Surgical Sterile Ophthalmic Pack,Disposable Opthalmic Surgical Pack Suzhou JaneE Medical Technology Co., Ltd. , https://www.janeemedical.com